Using methylprep from within an IDE¶
The primary methylprep API provides methods for the most common data processing and file retrieval functionality.
run_pipeline¶
Run the complete methylation processing pipeline for the given project directory, optionally exporting the results to file.
Returns: A collection of DataContainer objects for each processed sample
from methylprep import run_pipeline
data_containers = run_pipeline(data_dir, array_type=None, export=False, manifest_filepath=None, sample_sheet_filepath=None, sample_names=None)
| Argument | Type | Default | Description |
|---|---|---|---|
data_dir |
str, Path |
Base directory of the sample sheet and associated IDAT files | |
array_type |
str |
None |
Code of the array type being processed. Possible values are custom, 450k, epic, and epic+. If not provided, the pacakage will attempt to determine the array type based on the number of probes in the raw data. |
export |
bool |
False |
Whether to export the processed data to CSV |
manifest_filepath |
str, Path |
None |
File path for the array’s manifest file. If not provided, this file will be downloaded from a Life Epigenetics archive. |
sample_sheet_filepath |
str, Path |
None |
File path of the project’s sample sheet. If not provided, the package will try to find one based on the supplied data directory path. |
sample_names |
str collection |
None |
List of sample names to process. If provided, only those samples specified will be processed. Otherwise all samples found in the sample sheet will be processed. |
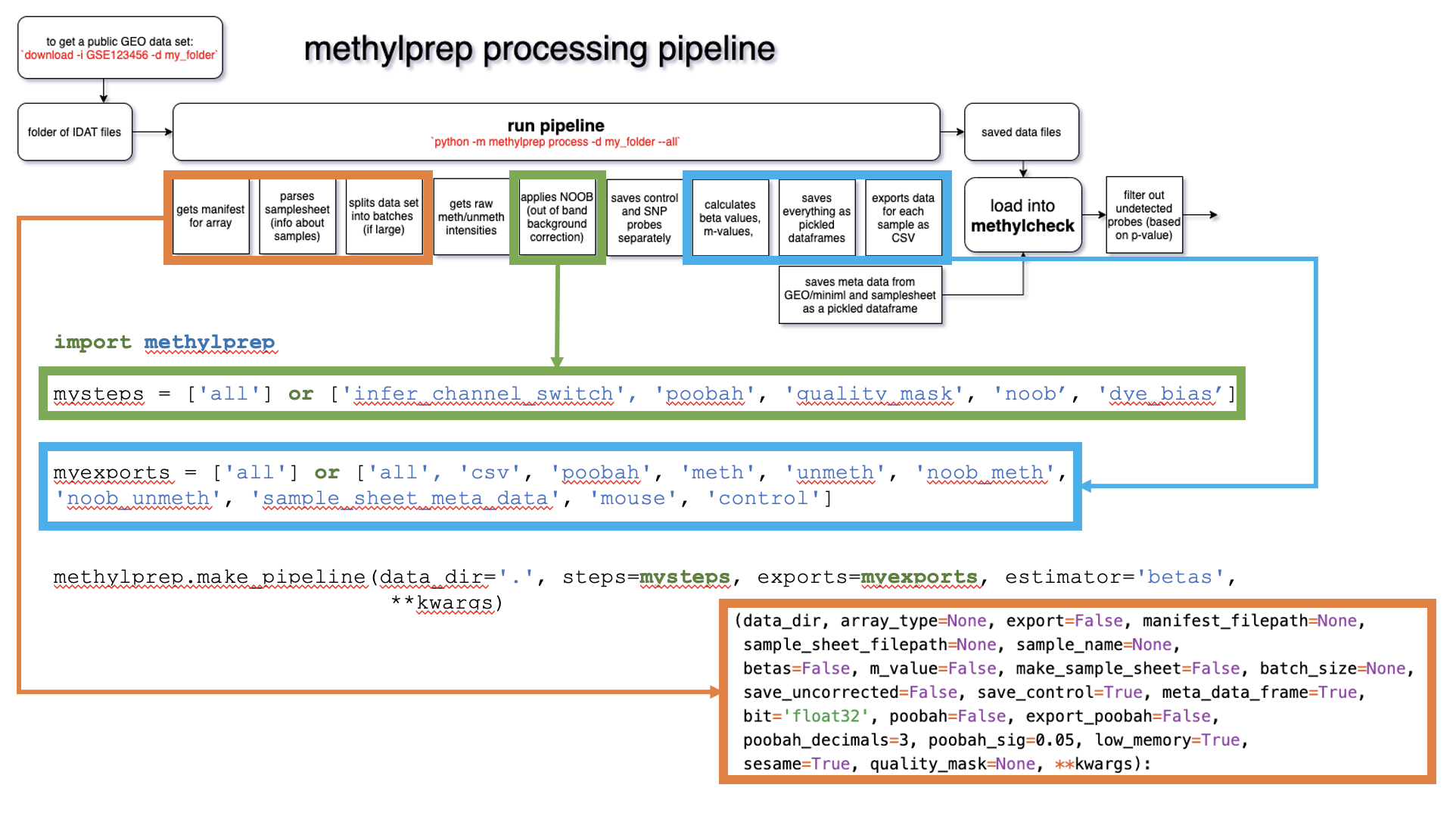
make_pipeline¶
This function works much like run_pipeline, but users are offered the flexibility to customize the pipeline to their liking. Any steps that they would like to skip may be excluded from the steps argument. Similarly, users may pick and choose what exports (if any) they would like to save.
| Argument | Type | Default | Description |
|---|---|---|---|
data_dir |
str, Path |
Base directory of the sample sheet and associated IDAT files | |
steps |
str, list |
None |
List of processing steps: [‘all’, ‘infer_channel_switch’, ‘poobah’, ‘quality_mask’, ‘noob’, ‘dye_bias’] |
exports |
str, list |
None |
file exports to be saved. Anything not specified is not saved. [‘all’, ‘csv’, ‘poobah’, ‘meth’, ‘unmeth’, ‘noob_meth’, ‘noob_unmeth’, ‘sample_sheet_meta_data’, ‘mouse’, ‘control’] |
estimator |
str |
beta |
what the pipeline should return [beta, m_value, copy_number, None] (returns data containers instead) |
If customizing the data processing steps interests you, you may also want to look at using the SampleDataContainer object, which is the output of processing when run in notebooks and beta_value or m_value is False. Each SampleDataContainer class object includes all of the sesame SigSet data sets and additional information about how the sample was processed.
get_sample_sheet¶
Find and parse the sample sheet for the provided project directory path.
Returns: A SampleSheet object containing the parsed sample information from the project’s sample sheet file
from methylprep import get_sample_sheet
sample_sheet = get_sample_sheet(dir_path, filepath=None)
| Argument | Type | Default | Description |
|---|---|---|---|
data_dir |
str, Path |
Base directory of the sample sheet and associated IDAT files | |
sample_sheet_filepath |
str, Path |
None |
File path of the project’s sample sheet. If not provided, the package will try to find one based on the supplied data directory path. |